
Understanding Bacteriophage Specificity
A quick synopsis of an interesting interaction experiment

Paper Review - Prediction of strain level phage–host interactions across the Escherichia genus using only genomic information (Oct. 2024)
The bacteriophage
My whole goal for this newsletter is to provide a corner of the internet to talk about bacteria, computation, and everything that comes between. Within that landscape, I’ve personally encountered a lot of folks who discuss and study the weird (but potentially ground-breaking) world of bacteriophages. These small robot-shaped little guys, for the uninitiated, are viruses with an insatiable desire to target bacteria and replicate using the bacteria’s genomic machinery against them. They achieve this level of specificity to bacteria by matching the surface proteins of bacterial species to the receptor proteins found on the tail end of the bacteriophage (henceforth I will shift between calling them phages and bacteriophages, just whatever feels good as I type).
This targeted specificity to certain bacteria offers a potential future use-case that can lead to profound changes in human health. For starters, the phage specificity can hopefully be engineered to target pathogenic bacteria (think Mycobacterium tuberculosis, the cause of Tuberculosis which causes a large share of deaths from infectious disease over the globe). Bacteria can gain the ability to evade antibiotics through random mutations to key genes, but with the addition of phages that can target the bacteria based on their surface proteins, this can lead to better therapeutics or treatments with high efficacy. Another potential use case is a bit more Sci-fi, but increased specificity and design of phages of targeted bacteria can lead to methods that can engineer microbiomes, something that is pretty tough to accomplish using current techniques.
Potential aside, there remains a large amount of work required to bring phage therapies/targeting to the mainstream. It turns out that engineering the specificity of phages to bacteria is a multi-step process that leads to exact pairings in an interaction being rather hard to predict or transfer between different bacterial species. These dynamics however could be elucidated through careful consideration of the bacterial and viral genomes and the features that they both possess. So, it begs the question, “How one can craft a methodology to tell which phages bind well for a certain bacteria?”.
Crafting a many-to-many dataset
One would imagine that in order to see what genomic features were indicative of phage binding to a bacterium, you first would need a diverse dataset of both bacteria and phages to compare against. Also (and this thought instantly makes me tired thinking about the amount of work this might have taken), you would need to do an unbiased and non-random pairing of bacteria and phages to see which interactions were successful while comparing to ones that had a minute effect. Together, having these two conditions met would lead to the ability to link genomic features to binding, unified through a predictive modeling framework!
The researchers of the paper being reviewed did this with a collection of 403 natural isolates belonging to the Escherichia genus and also 96 Escherichia-infecting phages (smartly done, to constrain the problem and deliver a good proof of concept). The combinatorically advantaged reader will discover that the number of unique combinations of phages and bacteria would total 38,688 which is even more staggering considering that there were 3 concentration levels and 3 replicates for each interaction.
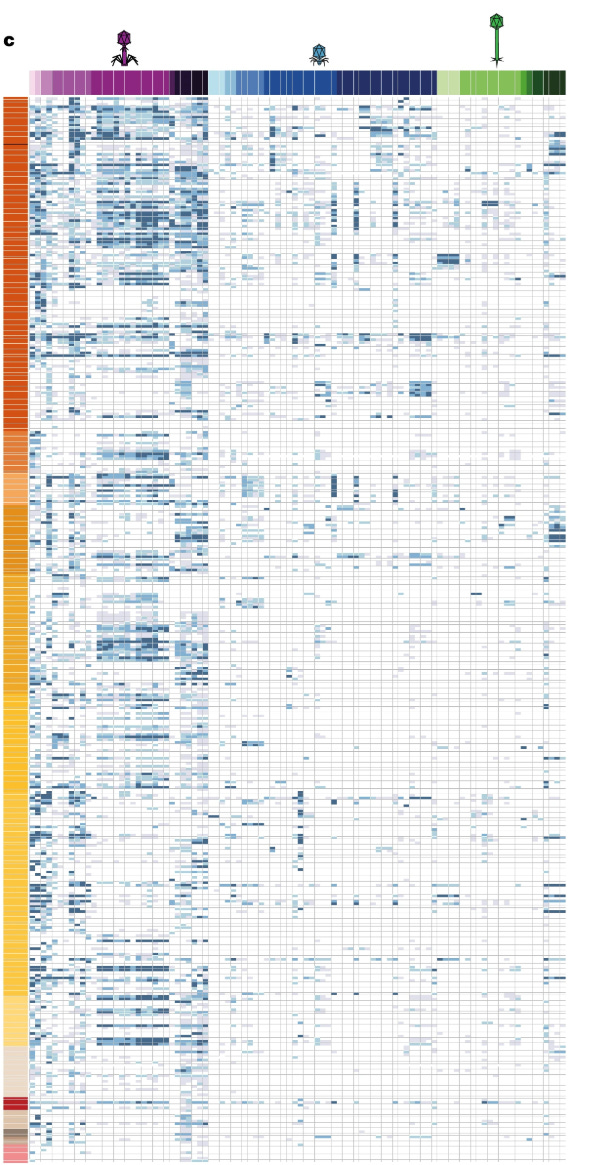
Here is the matrix of phage–bacteria interactions matrix. Rows represent the bacterial isolates while the columns represent a phage (Colors correspond to phage viral genera and bacterial phylogroups). Each cell of the matrix have a variable intensity of blues, which indicates the severity of infection a phage could enact on a bacteria.
Factors that influence binding to bacteria
The researchers used this intricate dataset as the foundation for a potential machine learning model that could account for phage specificity, but quickly realized the diversity of certain genomic factors and defense systems for phages led to a deluge of factors to account for in a putative model. Instead, the researchers looked to narrow down this large list of potential factors by quantifying their explanatory power.
The first interesting discovery the scientists made was that the phages that were found on the same isolation strain of bacteria were a major explanation for successful infection. Comparative genomics of the phage and its tail-spike & tail-fibre genes revealed remarkable conservation when the phages were coming from a bacteria belonging to the same strain. These two genes are hugely responsible for the phage Receptor Binding Protein (RBP), which is considered a hotspot for genomic variability in the phage. This suggests that the specificity of the RBP is a huge determinant of the infection potential of a phage, and furthermore that this conservation is based primarily on the diversity of isolation strains that can be procured from a particular bacteria. Additionally, on the bacteria side of things, the researchers found that surface proteins that impact adsorption also appeared to have a significant (but weak) impact on phage infections.
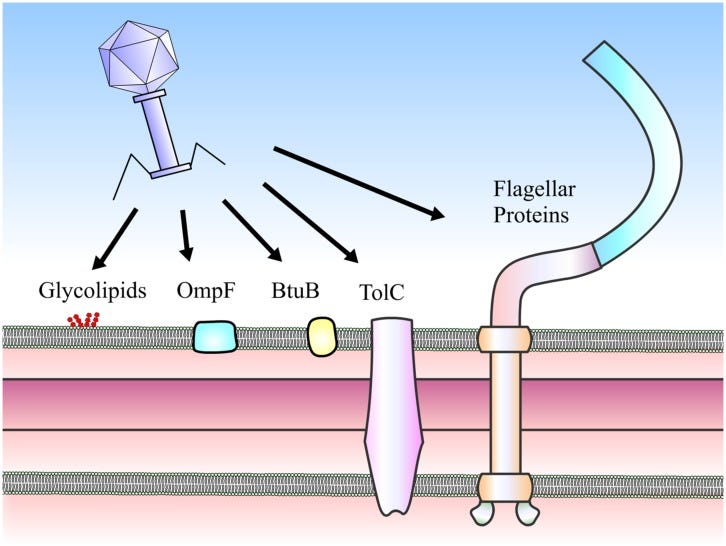
RBP is one of the stars of the show when it comes to viral entry to the bacteria. By recognizing a surface protein of interest, the bacteriophage achieves higher specificity in targeting the bacteria of interest. Picture comes from here.
Of interest, it turned out that even though bacteria possess a variety of anti-phage defense systems, these did little to predict the binding affinity of the phages to the bacteria. Surprising as the result may be, the researchers decided that the inclusion of these anti-phage defense systems was unnecessary for future predictive models.
After narrowing down the total number of features that could be used to infer interactions, the researchers were able to discover that their models that included only adsorption factors of the bacteria were able to achieve higher levels of discriminatory power than considering the isolated strain the phages were found on. The best model that was crafted reported an AUROC of 86% (this is considered to be excellent in model performance!) after 10-fold cross-validation.
This is a promising first step in being able to predict these complex interactions, but false positives/negatives still were present amongst the classifications. This is my own personal conjecture, but I think the reason might be because of the assumptions that the data collected might enforce. For instance, a researcher might have a personal assumption (myself included) that most of these interactions can be fully explained by genomic variability, so they only collect data associated with the genomes of the bacteria and phages. Imagine the things that aren’t collected here such as abiotic factors that would also be crucial to getting fine-tuned control, but are outside of the researcher’s control. I’m sure this will be addressed in future studies.
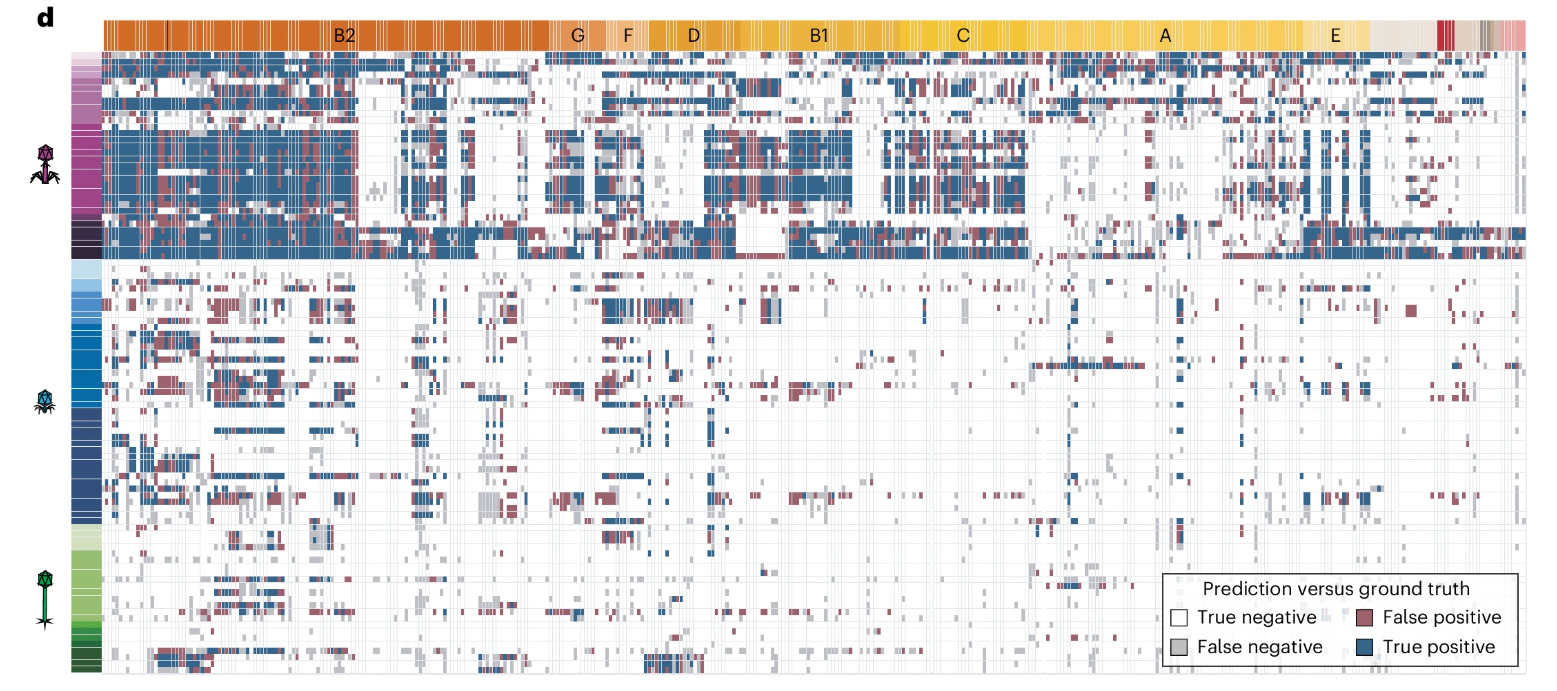
The model the researchers developed was used to assess the predictive ability of a successful interaction between a phage and it’s target. The white and blue cells in this heatmap highlight where the model was correct in deciphering the interaction while gray and red cells are where the model fell short of classifying properly. I think a further analysis of what led the model to make false classification given the input data supplied might suggest the fuzziness of what these features can tell us about their usefulness in correct classification.
The need for the ‘all vs. all’ study
The reason I wanted to read and discuss a bit about this research is because of the scale of the work - a full dataset of bacteria/phage interactions that were also graded on the severity of the interaction is the type of experiment you can only dream about! In a future newsletter, I am considering focusing more on biology experiments that leverage ‘all vs. all’ strategies to understand what factors contribute to successful and unsuccessful interactions. I think the thing that excites me about this study however is that there is room here one day to exploit the random pairings of genomically diverse bacteria with genomically diverse phages. In the current research article, the problem was constrained to a particular genus of bacteria, but I think it will be necessary to develop randomly diverse libraries of bacteria that truly probe which genomic features confer phage binding affinity.
I’m under no sanguine idea that this would be easy, and in fact, would lead to a plethora of data that might come out as sparse and essentially not viable. However, minimal success could be built upon, and specific machine learning models will get better at figuring out the genomic features that make bacteriophages so lethal. It’s pretty close to happening given the right type of experiment and resources - or at least a guy can dream about it.
In closing
I only presented a snapshot of this expansive paper; I encourage everyone to give it a closer read.
Continuously dividing,
N.L.